Abstract:
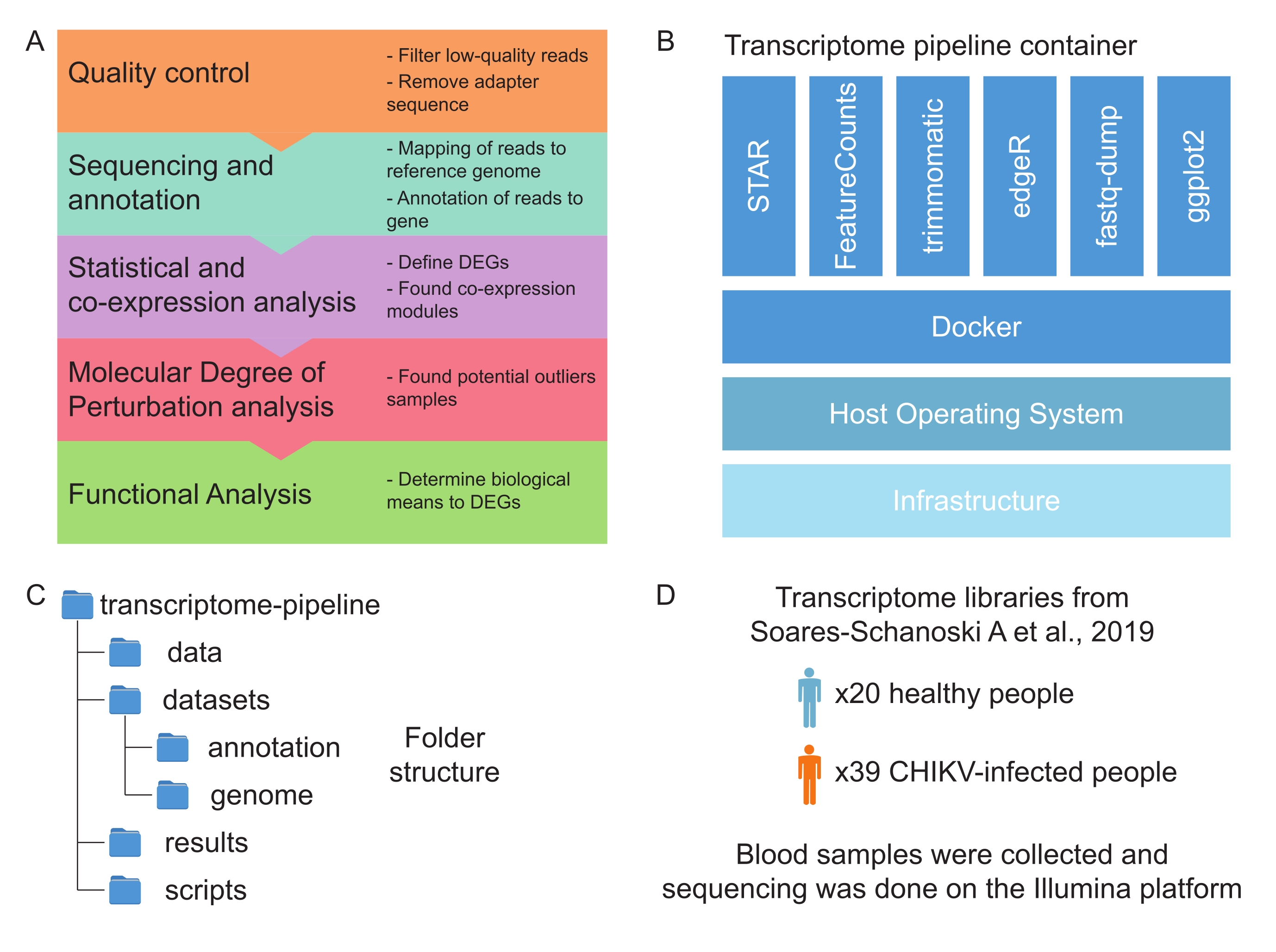
Pathogens can cause a wide variety of infectious diseases. The biological processes induced by the host in response to infection determine the severity of the disease. To study such processes, researchers can use high-throughput sequencing techniques (RNA-seq) that measure the dynamic changes of the host transcriptome at different stages of infection, clinical outcomes, or disease severity.This investigation can lead to a better understanding of the diseases, as well as uncovering potential drug targets and treatments. The protocol presented here describes a complete pipeline to analyze RNA-sequencing data from raw reads to functional analysis. The pipeline is divided into five steps: (1) quality control of the data; (2) mapping and annotation of genes; (3) statistical analysis to identify differentially expressed genes and co-expressed genes; (4) determination of the molecular degree of the perturbation of samples; and (5) functional analysis. Step 1 removes technical artifacts that may impact the quality of downstream analyses. In step 2, genes are mapped and annotated according to standard library protocols. The statistical analysis in step 3 identifies genes that are differentially expressed or co-expressed in infected samples, in comparison with non-infected ones. Sample variability and the presence of potential biological outliers are verified using the molecular degree of perturbation approach in step 4. Finally, the functional analysis in step 5 reveals the pathways associated with the disease phenotype. The presented pipeline aims to support researchers through the RNA-seq data analysis from host-pathogen interaction studies and drive future in vitro or in vivo experiments, that are essential to understand the molecular mechanism of infections.
Figure: